

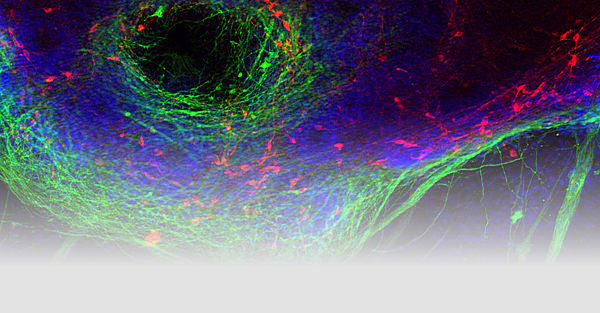
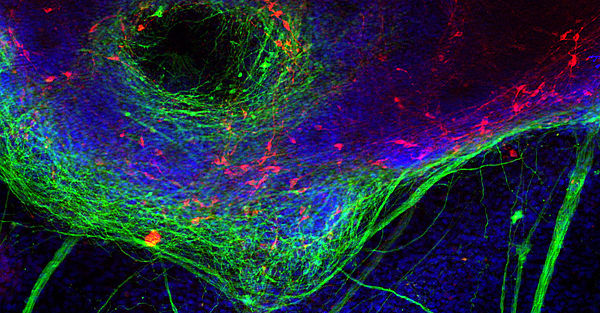
Age-related neurodegenerative diseases are a severe and increasingly worrisome burden for our aging population. Most of the chronic neurodegenerative diseases (Parkinson’s disease [PD], Lewy body dementia [LBD], Alzheimer’s disease, frontotemporal dementia [FTD], amyotrophic lateral sclerosis [ALS], etc.) are characterized by intracellular protein inclusions that are specific for each of these diseases. We investigate the structural, molecular, cellular, and histopathological mechanisms underlying aggregation of the PD/DLB-associated synaptic protein α-synuclein as well as the FTD/ALS-associated nucleic acid binding proteins TDP-43 and FUS/TLS. Pathological pathways modelled in cell culture and animal models (mice and flies). Investigated mechanisms include protein aggregation and phosphorylation, neuroinflammation, mitophagy, and nuclear import. We wish to understand the molecular basis of the remarkable specificity of intracellular protein aggregation killing particular neuronal subpopulations, which cause the characteristic syndromes of neurodegenerative movement disorders and dementias.
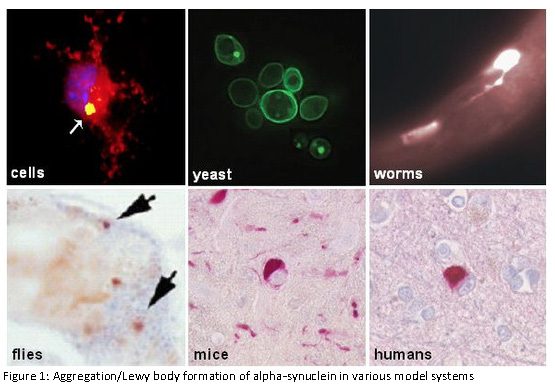
The research group is funded by the Hertie-Institute and the German Center for Neurodegenerative Diseases.
Characterization and Behavioural Consequences of α-Synucleinopathy in Transgenic Mice
α-Synuclein is one of the top-most genetic risk factors for Parkinson's disease (PD) and the protein is the major building block of Lewy bodies, the neuropathological hallmarks in the brain of PD patients. We are using transgenic mice expressing human mutant A30P α-synuclein under the control of a Thy1 promoter, which recapitulate human α-synucleinopathy down to the ultrastructural level. Cognitive behavior of (Thy1)-h[A30P]αSYN mice is impaired in an age-dependent manner, most likely due to development of neuropathology and neuronal dysfunction within the amygdala circuitry (Schell et al. 2012). Moreover, old transgenic mice ultimately die of locomotor deterioration, caused by brain stem and spinal motoneuron pathology. The age of onset of this terminal phenotype is accelerated by high fat diet-induced obesity (Rotermund et al. 2014). We are further investigating epigenetic mechanisms influenced by α-synuclein in cell culture and in vivo (Sugeno et al. 2016). This work is supported by the German Center for Neurodegenerative Diseases (DZNE) and the Transnational German-Canadian-French epigenomics consortium DecipherPD.
Regulation of leucine-rich repeat kinase 2 in macrophages
Missense mutations in the gene encoding leucine-rich repeat kinase 2 (LRRK2) are the most common cause of autosomal-dominant hereditary PD and a considerable genetic risk factor for sporadic PD. However, little is known about the molecular mechanisms of LRRK2 activation, and the biological role of LRRK2 is still largely unknown. Interestingly, a large amount of LRRK2 is expressed in the immune system. We found that LRRK2 induction by interferon-g in macrophages is mediated via a signal transduction pathway dependent on the extracellular signal-regulated kinase 5 (Kuss et al. 2014). We are currently investigating the cellular functions of LRRK2 in stimulated macrophages. This work is supported by the Michael J. Fox Foundation.
Regulation and Cellular Effects of Parkin E3 Ubiquitin Ligase Activities
Most of the familial PD cases are caused by recessive mutations in the PARK2/PARKIN gene, which may also be a genetic risk factor for sporadic PD. The PARKIN gene product functions as an E3 ubiquitin protein ligase for a variety of unrelated substrate proteins. We investigate the role of parkin in the autophagic degradation of damaged mitochondria. Parkin is recruited to experimentally depolarized mitochondria in a PINK1-dependent manner, which is differentially affected by PD mutations in both genes (Geisler et al. 2010). Parkin ubiquitinylates a distinct set of mitochondrial outer membrane protein in a highly complex manner. We identified a set of E2 ubiquitin-conjugating enzymes that mediate such reactions (Geisler et al. 2014). We are further investigating the complex regulation of PINK1/parkin-regulated mitophagy, also using high content imaging. This work is supported by the German Center for Neurodegenerative Diseases).
Cell Biology of the FTD/ALS Associated Nuclear Splice Factors TDP-43 and FUS
TDP-43 and FUS/TLS in cytosolic and nuclear inclusions are neuropathological hallmarks of frontotemporal dementia (FTD) and amyotrophic lateral sklerosis (ALS). It remains to be elucidated if the cytosolic aggregates are actively neurotoxic or if cytosolic sequestration of these nuclear proteins deprives neurons of vital RNA processing factors. To identify novel target genes, we conducted expression profiling studies after RNA interference. We have identified the intracellular transport protein histone deacetylase 6 and the exon junction complex component SKAR as novel TDP-43 target mRNAs and validated them structurally and functionally in non-neuronal and neuronal cells treated with siRNA and lentiviral shRNA vectors as well as in TDP-43 mutant animal models (Fiesel et al. 2010 and 2011). We unraveled the regulation of ubiquitinylation of TDP-43 by UBE3E ubiquitin-conjugating enzymes and the ubiquitin isopeptidase UBPY, and studied the effects on TDP-43 aggregation and neurotoxicity in Drosophila (Hans et al. 2014). We are currently investigating more systematically post-translational modifications of TDP-43 with a focus on lysine-ubiquitinylations and -acetylations. For FUS we could demonstrate in Drosophila modifier effects of the nuclear import regulatory proteins transportin and protein arginine methyltransferase 1 (Jäckel et al. 2015). This work is supported by the German Research Council (DFG), the German Center for Neurodegenerative Disease (DZNE) and the NOMIS Foundation.

+49 (0)7071
29-81968
+49 (0)7071
29-81968

07071
29-81970
+49 (0)7071
29-81968
+49 (0)7071-
29-81968
Selected publications
Jäckel, S., Summerer, A. K., Thömmes, C. M., Pan, X., Voigt, A., Schulz, J. B., Rasse, T. M., Dormann, D., Haass, C., and Kahle, P. J. (2015) Nuclear import factor transportin amd arginine methyltransferase 1 modify FUS neurotoxicity in Drosophila. Neurobiol. Dis. 74, 76-88
Rotermund, C., Truckenmüller, F. M., Schell, H., and Kahle, P. J. (2014) Diet-induced obesity accelerates the onset of terminal phenotypes in a-synuclein transgenic mice. J. Neurochem. 131, 848-858
Geisler, S., Vollmer, S., Golombek, S., and Kahle, P. J. (2014) UBE2N, UBE2L3 and UBE2D2/3 ubiquitin-conjugating enzymes are essential for parkin-dependent mitophagy. J. Cell Sci. 127, 3280-3293
Hans, F., Fiesel, F. C., Strong, J. C., Jäckel, S., Rasse, T. M., Geisler, S., Springer, W., Schulz, J. B., Voigt, A., and Kahle, P. J. (2014) UBE2E ubiquitin-conjugating enzymes and ubiquitin isopeptidase Y regulate TDP-43 protein ubiquitination. J. Biol. Chem. 289, 19164-19179
Kuss, M., Adamopoulou, E., and Kahle, P. J. (2014) Interferon-gamma induces leucine-rich repeat kinase LRRK2 via extracellular signal-regulated kinase ERK5 in macrophages. J. Neurochem. 129, 980-987
Schell, H., Boden, C., Maia Chagas, A., and Kahle, P. J. (2012) Impaired c-Fos and polo-like kinase 2 induction in the limbic system of fear-conditioned alpha-synuclein transgenic mice. PLoS ONE 7, e50245
Fiesel, F. C., Weber, S. S., Supper, J., Zell, A., and Kahle, P. J. (2011) TDP-43 regulates global translational yield by splicing of exon junction complex component SKAR. Nucleic Acids Res. 40, 2668-2682
Geisler, S., Holmström, K. M., Treis, A., Skujat, D., Weber, S. S., Fiesel, F. C., Kahle, P. J., and Springer, W. (2010) The PINK1/parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 6, 871-878
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., and Springer, W. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119-131
Fiesel, F. C., Voigt, A., Weber, S. S., Van den Heute, C., Waldenmaier, A., Görner, K., Walter, M., Anderson, M. L., Kern, J. V., Rasse, T. M., Schmidt, T., Springer, W., Kirchner, R., Bonin, M., Neumann, M., Baekelandt, V., Alunni-Fabbroni, M., Schulz, J. B., and Kahle, P. J. (2010) Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J. 29, 209-221





