

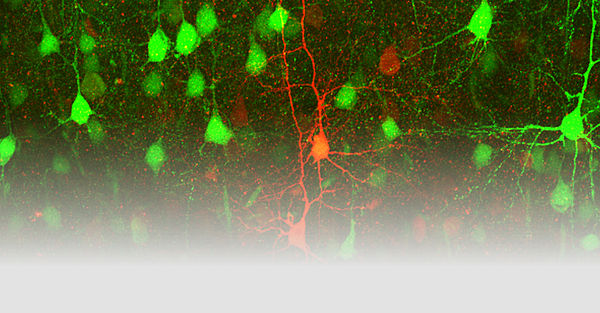
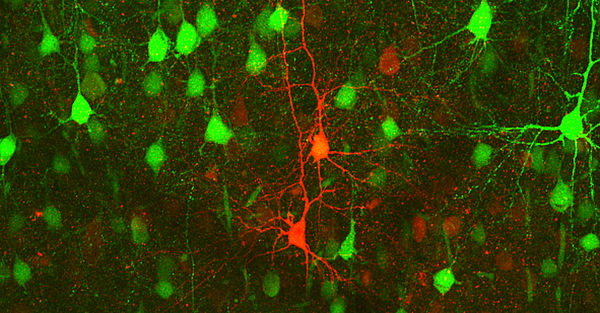
Our research group uses transgenic mouse models to investigate the molecular mechanisms of disease-associated variants in genes encoding neuronal ion channels. These so-called channelopathies include epilepsies and migraine, two of the most common neurological diseases.
Epileptic seizures result from spontaneous, synchronous electrical activity of groups of neurons in the brain, manifesting in a wide range of clinical symptoms, such as tingling in an extremity to impaired consciousness, complex actions, or generalized seizures. Migraine attacks often begin with an aura, which classically manifests as visual complaints. These auras are due to cortical spreading depolarization (CSD), a wave of neuronal depolarization that travels slowly across the cerebral cortex. Thus, both diseases are associated with pathological electrical activity, epilepsy with a fast, migraine with a slow propagation of abnormal activity. Interestingly, defects in the same gene can cause epilepsy and migraine, and both diseases can also occur in the same patient and trigger each other.
In our projects, we aim to understand the relationship between genetic variants and rare ion channel diseases at the molecular, cellular and network level and the underlying mechanism of hyperexcitability of these diseases (epileptic seizures vs. CSD) in order to develop new therapies. For this purpose, we use various electrophysiological and optical methods, which are complemented by transcriptome studies. The rare ion channel diseases also provide a model for the pathophysiology and therapy of much more common sporadic diseases with similar symptoms.
Epileptogenesis of Genetic Epilepsies
The goal of the research group FOR-2715 "Epileptogenesis of Genetic Epilepsies" is to elucidate whether and how genetic variants trigger a cascade of epileptogenic events and how these interact with brain development. In our project, we aim to understand the epileptogenic mechanisms of KCNA2-associated encephalopathies. For this, we use in utero electroporation of KCNA2 variants as well as newly developed knock-in mouse models with gain- or loss-of-function variants. Using these mouse models, we aim to investigate the vulnerable time window of Kcna2 variants and network dysfunction of the developing neuronal network. In addition, we are looking into the treatment of patients with KCNA2 gain-of-function variants with 4-aminopyridine and are currently developing Kcna2 antisense oligonucleotides as a new therapeutic option.
Treat-ION
In the Treat-ION research network, we focus on the pathophysiological mechanisms of hemiplegic migraine (HM), a severe monogenic subtype of migraine with some degree of hemiplegic motor weakness during the aura. Here, we aim to investigate the pathophysiological mechanisms of epileptic seizures and cortical spreading depolarization caused by variants in the SLC1A3 gene encoding the glutamate transporter and anion channel EAAT1. In addition, we aim to develop novel and individualized therapies for patients with this rare disease.
Gruppo Famiglie Dravet Associazione ONLUS
In another project funded by Gruppo Famiglie Dravet Associazione ONLUS in partnership with other European Dravet foundations, we are interested in a new mechanism in the pathophysiology of Dravet syndrome (DS), namely the interactions between neurons and oligodendrocytes. This disease is mostly caused by genetic variants in the SCN1A gene, which encodes the NaV1.1 channel, the major Na+ channel in inhibitory neurons. Here, we investigate the effects of SCN1A variants in neurons, oligodendrocyte progenitor cells (OPCs), and oligodendrocytes of a DS mouse model, as these cells all express the NaV1.1 channel, share a common embryonic origin, and interact strongly during development.
European Joint Program on Rare Disease (EJP RD)
As part of the European Joint Program on Rare Disease (EJP RD), we are focusing on targeted treatments for Dravet syndrome using knock-in animal models as well as human disease models as part of the nationally funded (by DFG for Germany) consortium SCN1A-UP! with partners in France, Italy, Belgium, the Netherlands and Germany.

+49 (0) 7071
29-81984

+49 (0) 7071
2981914

+49 (0) 7071
29-80440

+49 (0) 7071
29- 81968
+49 (0)7071
29-81921
You can find the complete list of publication on ORCID
Hedrich UBS*, Lauxmann S*, Wolff M, …, Schwarz N, Fudali M, Lerche H. Effective precision therapy in KCNA2-related developmental and epileptic encephalopathy with 4-aminopyridine, Sci Trans Med 2021 Sep;13(609):eaaz4957, *equal contribution.
Auffenberg E*, Hedrich UBS*, Barbieri R*, Miely D*, Groschup B, Wuttke TV, Vogel N, Lührs P, Zanardi I, Bertelli S, Spielmann N, Gailus-Durner V, Fuchs H, Hrabe de Angelis M, Pusch M, Dichgans M, Lerche H, Gavazzo P§, Plesnila N§, Freilinger T§. Hyperexcitable interneurons trigger cortical spreading depression in an Scn1a migraine-model, JCI 2021 Nov 1;131(21):e142202. *equal contribution.
Lauxmann S, Verbeek NE, Liu Y, Zaichuk M, Müller S, Lemke JR, van Kempen MJA, Lerche H, Hedrich UBS. Relationship of electrophysiological dysfunction and clinical severity in SCN2A-related epilepsies. Hum Mutat 2018;39:1942-1956.
Masnada S*, Hedrich UBS*, Gardella E, Schubert J, Kaiwar C, Klee EW, Lanpher BC, Gavrilova RH, Synofzik M, Bast T, …, Lerche H, Rubboli G. Clinical spectrum and genotype–phenotype associations of KCNA2-related encephalopathies. Brain 2017;140(9):2337-54, *equal contribution.
Wolff M*, Johannesen KM*, Hedrich UB*, Masnada S, Rubboli G, Gardella E, Lesca G, Ville D, Milh M, Villard L, Afenjar A, Chantot-Bastaraud S, Mignot C, Lardennois C, Nava C, Schwarz N, …, Kluger G, Lerche H & Moller RS. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017; * equal contribution.
Schwarz N, Hahn A, Bast T, Muller S, Loffler H, Maljevic S, Gaily E, Prehl I, Biskup S, Joensuu T, Lehesjoki AE, Neubauer BA, Lerche H & Hedrich UB. Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J Neurol 2016;263:334-43.
Syrbe S*, Hedrich UB*, Riesch E*, Djemie T*, Muller S, Moller RS, Maher B, Hernandez-Hernandez L, Synofzik M, Caglayan HS, Arslan M, Serratosa JM, Nothnagel M, May P, Krause R, Loffler H, Detert K, Dorn T, Vogt H, Kramer G, Schols L, Mullis PE, Linnankivi T, Lehesjoki AE, Sterbova K, Craiu DC, Hoffman-Zacharska D, Korff CM, Weber YG, Steinlin M, Gallati S, Bertsche A, Bernhard MK, Merkenschlager A, Kiess W, Euro ER, Gonzalez M, Zuchner S, Palotie A, Suls A, De Jonghe P, Helbig I, Biskup S, Wolff M, Maljevic S, Schule R, Sisodiya SM, Weckhuysen S, Lerche H, Lemke JR. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat Genet 2015;47:393-9 * equal contribution.
Hedrich UBS, Liautard C, Kirschenbaum D, Pofahl M, Lavigne J, Liu Y, Theiss S, Slotta J, Escayg A, Dihne´M, Beck H, Mantegazza M, and Lerche H. Impaired Action Potential Initiation in GABAergic Interneurons Causes Hyperexcitable Networks in an Epileptic Mouse Model Carrying a Human NaV1.1





