

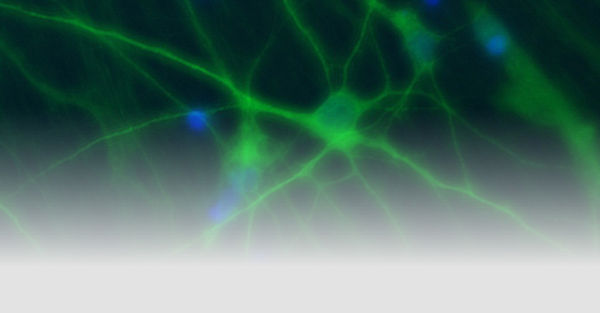
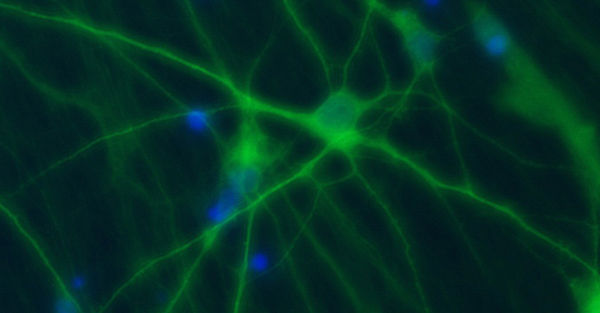
The goal of our research is to link the molecular mechanisms of mainly genetic, neurological diseases caused by disturbed neuronal excitability to their clinical symptoms and a personalized treatment. We are recruiting well-defined cohorts of patients with epilepsies and related disorders, searching for disease-causing genetic defects with modern sequencing techniques, particularly in ion channels or transporters, and analyzing their functional consequences to understand the pathomechanisms. A particular focus is
on finding and exploring new personalized therapies for genetic disorders. To study mechanisms of neuronal hyperexcitability on the molecular, cellular and network level, we use non-neuronal screening tools such as automated electrophysiology in oocytes and mammalian cells, neuronal expression systems including neurons derived from induced pluripotent stem cells and human brain slices, and gene-targeted mouse models.
- Functional investigations of genetic defects in ion channels
With the BMBF-funded Treat-ION consortium on Neurological Ion Channel and Transporter Disorders we focus on therapeutic studies in cellular, animal and human models, which are complemented by in silico searches for new treatments, better predictions for the functional consequences of mutations for therapeutic purposes and cellular drug screens. The use of approved and available ‘repurposed’ drugs such as 4-aminopyridine is a a specific goal to enable precision treatment. Our findings are directly delivered to patients through molecular therapeutic boards attached to the German academy of rare neurological diseases (DASNE) and the centers for rare diseases (ZSEs) in Baden-Württemberg and through a structured process for drug repurposing. Functional implications of selected mutations are examined in neuronal expression systems, such as transfected murine primary neurons, in utero electroporated neurons and genetically altered animal models carrying a human mutation (so-called “humanized mouse models”). The advantage of both in utero electroporated neurons and gene-targeted mouse models is that altered channels can be studied in their natural environment and additionally, the consequences on intrinsic neuronal properties and network activity can be studied using single cell patch clamp, extracellular recording or multielectrode array (MEA) techniques. We perform 256 electrode MEA recordings and high-resolution electrical imaging (CMOS with 4000 electrodes) to analyze single cell compartments and neuronal network activity in brain slices of transgenic animals and study network dysfunction of our mouse models in vivo together with O. Garaschuk (Inst. Neurophysiology) using Ca2+ imaging in the frame of the DFG Research Unit. To gain insight into the exact mechanisms as to how epilepsy develops as a consequence of a genetic defect, we investigate brain region- and time-specific RNA expression using single cell RNA sequencing in distinct neuronal subpopulations in mouse models.
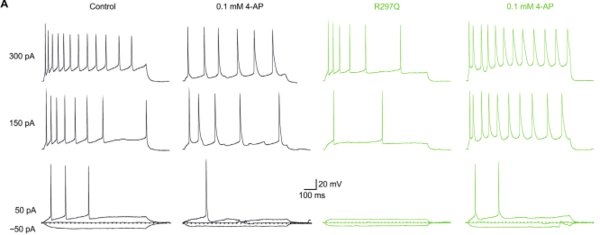
Two murine neurons recorded with the patch clamp technique before and after appliaction of 100 µM 4-AP. In black a KCNA2 wildtype transduced neuron and in green a KCNA2 gain of function transduced neuron. - Induced pluripotent stem cells as epilepsy models
Since human brain tissue is a limited resource in neuroscience research, we are additionally generating neurons from induced pluripotent stem cells (iPSCs). These represent an attractive, albeit simplified, alternative of a human model system to study diseases of the nervous system.
By reprogramming patient-specific fibroblasts into pluripotent stem cells and subsequently overexpressing specific transcription factors, we can specifically generate excitatory and inhibitory cortical neurons. In subsequent functional studies, the disease mechanism of individual patients can be investigated taking into account their unique genetic background. We investigate network activity using a multiwell-MEA system as well as single cell activity by patch clamp measurements. By this, we were already able to show that developmental electrophysiological activity patterns in iPSC-derived neurons are comparable to those in humans and animals (Rosa et al., Stem Cell Rep 2020). Current projects include electrophysiological analysis of patient-specific iPSC neurons carrying pathological mutations in genes encoding ion channels (KCNA2, KCNH1) or synaptic proteins (STX1B).
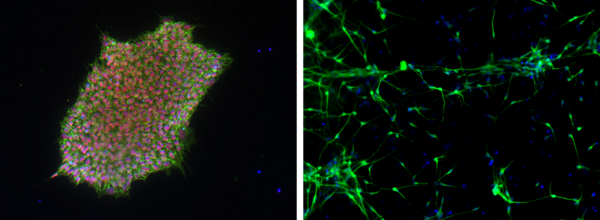
Expression of the markers SOX2 (green) and SEEA4 (red) indicates pluripotency of iPSCs (left). iPSC-derived neurons, two weeks after spontaneous embryoid body differentiation (right). - Human brain slices cultures
As another human model system, we are using human slice cultures which can be maintained for up to four weeks with good neurophysiological properties when human cerebrospinal fluid (CSF) is used as culture medium. The use of ex vivo brain slices derived from adult human neurosurgical-resected tissue allows to probe electrophysiological properties at single cell and network level (Schwarz et al., Sci Rep 2017). We demonstrated robust preservation of neuronal cytoarchitecture and electrophysiological properties of human pyramidal neurons. Further experiments delineate the optimal conditions for efficient viral transduction of cultures, enabling ‘high throughput’ fluorescence-mediated 3D reconstruction of genetically targeted neurons, and demonstrate feasibility of long term live cell imaging of human cells in vitro.

Excess brain tissue from neurosurgical operations is cut into small sections (left) and cultured for weeks. The neurons retain their special morphology (right).

+49 (0) 7071
2-981983
+49 (0)7071-
29-81914

+49 (0) 7071
29-80442
+49(0)7071-
29-81914
+49 (0)7071-
29-81914

+49 (0)7071-
29-81914
+49 (0) 7071
29-87639
+49 (0)7071
29 81914

+49 (0) 7071
2981914

+49(0)7071-
29-85247

+49 (0) 7071
29-87638

+49 (0)7071-
29-81921
+49 (0)7071
29-81983

+49 (0) 7071
29-81984
+49 (0)7071-
29-81921
+49 (0) 7071
29-86525

+49 (0) 7071
2981914
+49 (0)7071-
29-87638

+49 (0) 7071
29-86588
+49 (0) 7071
29-80442
+49 (0)7071-
29-81921

+49 (0)7071-
29-81983

+49 (0) 7071
29-80442

+49 (0) 7071
29-81921

+49 (0) 7071
29-81922
+49 (0) 7071
29-80440

+49 (0) 7071
29-80440
+49 (0) 7071
29- 81968
+49 (0)7071
29-81914

+49(0)7071
29- 80442

+49 (0) 7071-
29-81914
+49 (0)7071
29-81921
+49 (0) 7071-
29 81914
+49 (0)7071-
29-80440
+49 (0) 7071
29-81914
Selected Publications
Müller P*, Takacs DS*, Hedrich UBS, Coorg R, Masters L, Glinton KE, Dai H, Cokley JA, Riviello JJ, Lerche H#, Cooper EC#. KCNA1 gain-of-function epileptic encephalopathy treated with 4-aminopyridine. Ann Clin Transl Neurol. 2023 Feb 15.
Krüger J, Schubert J, Kegele J, Labalme A, Mao M, Heighway J, Seebohm G, Yan P, Koko M, Aslan-Kara K, Caglayan H, Steinhoff BJ, Weber YG, Keo-Kosal P, Berkovic SF, Hildebrand MS, Petrou S, Krause R, May P, Lesca G, Maljevic S, Lerche H. Loss-of-function variants in the KCNQ5 gene are implicated in genetic generalized epilepsies. EBioMedicine. 2022 Oct;84:104244. doi: 10.1016/j.ebiom.2022.104244.
Johannesen KM*, Liu Y*, Koko M, Gjerulfsen CE, Sonnenberg L, Schubert J, Fenger CD, Eltokhi A, Rannap M, Koch NA,Lauxmann S, Krüger J, Kegele J, (…),Hedrich UBS, Benda J, Gardella E,Lerche H#, Møller RS#. Genotype-phenotype correlations in SCN8A-related disorders reveal prognostic and therapeutic implications. Brain 2021:awab321.
Auffenberg E*, Hedrich UB*, Barbieri R*, Miely D*, Groschup B, Wuttke TV, (…), Pusch M, Dichgans M, Lerche H, Gavazzo P#, Plesnila N#, Freilinger T#. Hyperexcitable interneurons trigger cortical spreading depression in an Scn1a migraine model. J Clin Invest 2021;131:e142202.
Hedrich UBS*, Lauxmann S*, (…), Bosselmann C, Schwarz N, Fudali M, Lerche H. 4-Aminopyridine is a promising treatment option for patients with gain-of-function KCNA2-encephalopathy. Sci Transl Med 2021;13:eaaz4957.
Koko M, Krause R, Sander T, Bobbili DR, Nothnagel M, May P,Lerche H; Epi25 Collaborative. Distinct gene-set burden patterns underlie common generalized and focal epilepsies. EBioMedicine 2021;72:103588.
Rosa F, Dhingra A, Uysal B, Mendis GDC, Loeffler H, Elsen G, Mueller S, Schwarz N, Castillo-Lizardo M, Cuddy C, Becker F, Heutink P, Reid CA, Petrou S, Lerche H#, Maljevic S#. In Vitro Differentiated Human Stem Cell-Derived Neurons Reproduce Synaptic Synchronicity Arising during Neurodevelopment. Stem Cell Reports 2020;15:22-37.
Schwarz N, Hedrich UBS, Schwarz H, P A H, Dammeier N, Auffenberg E, Bedogni F, Honegger JB, Lerche H, Wuttke TV, Koch H. Human Cerebrospinal fluid promotes long-term neuronal viability and network function in human neocortical organotypic brain slice cultures. Sci Rep. 2017 Sep 25;7(1):12249.

Hertie Center of Neurology
Hertie Institute for Clinical Brain Research
Department Neurology and Epileptology
Hoppe-Seyler-Straße 3
72076 Tübingen
Phone: +49 (0)7071 29-80442
Fax: +49 (0)7071 29-4488
Sabrina Kreiser
Yvonne Brändle
Phone: +49 (0)7071 29-80442
Fax: +49 (0)7071 29-4488
sekretariatne5.HL@med.uni-tuebingen.de
Heidrun Löffler
Phone: +49 (0)7071 29-81922
heidi.loeffler@medizin.uni-tuebingen.de




