

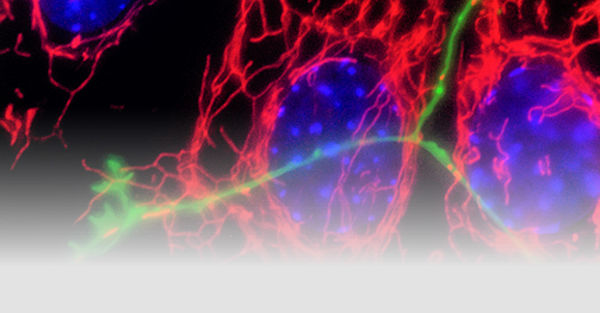
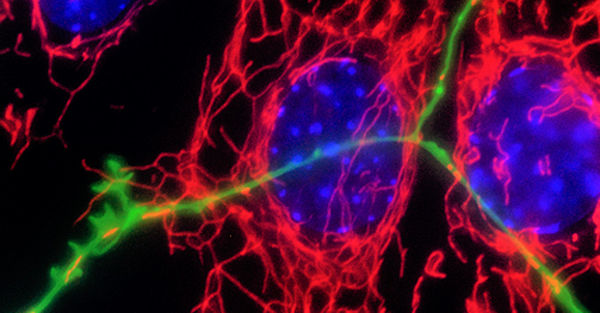
Ein Teil von Fällen neurodegenerativer Erkrankungen ist mit einem genetischen Risiko und Genmutationen verbunden. Jedoch sind die meisten Fälle von Morbus Parkinson sporadisch und die Erkrankung ist insgesamt sehr heterogen im Bezug auf Symptome und Pathologie. Die AG Fitzgerald hat sich zum Ziel gesetzt, die molekularen Mechanismen, welche der Neurogeneration zugrunde liegen, zu verstehen, indem genetische Formen der Erkrankung genutzt werden, um einen Ansatz für die Modellierung der Krankheit zu erlagen.
Mitochondrien sind wichtige Organellen, welche Energie produzieren und viele andere Funktionen besitzen, die für den zentralen Metabolismus und Zellsignaling benötigt werden. Neurone verwenden viel Energie und verlassen sich auf die engmaschige Kontrolle für Effizienz und die Reduktion der oxidativen Last. Mitochondriale Dysfunktion ist ein Phänomen, welches alle neurodegenerativen Erkrankungen betrifft und wichtig sein dafür sein könnte, die selektive Anfälligkeit bestimmter Hirnzellen zu erklären. Das ist besonders wichtig für Morbus Parkinson, da bei dieser Erkrankung dopaminerge Neurone aufgrund dessen sterben, dass Mitochondrien eine Hauptquelle für oxidativen Stress sind und zudem Dopamin degradieren.
Die Verbindung zwischen mitochondrialer Dysfunktion und Erkrankung in Morbus Parkinson wurde dadurch bewiesen, dass Umweltfaktoren und Krankheitsgene identifiziert wurden, welche im Besonderen die Mitochondrien betreffen. Diese Erkenntnisse wurden dazu verwendet, um viel Arbeit in die Aufklärung der Rolle der mitochondrialen Dysfunktion in Morbus Parkinson zu investieren. Allerdings sind die genauen Mechanismen, welche sporadischen Formen von Morbus Parkinson zugrunde liegen, weniger genau definiert.
Wir verwenden mehrere verschiedene Zellmodelle mit einem Fokus auf Zellen, welche von Patienten stammen. In enger Zusammenarbeit mit der Biodatenbank des Hertie-Instituts und der neurologischen Klinik haben wir gegenwärtig primäre humane Fibroblasten in Kultur, welche von Patienten und gesunden Individuen stammen. Außerdem haben wir induzierte pluripotente Stammzellen (iPSCs), neuronale Vorläuferzellen (smNPCs) und reife dopaminerge Neurone, welche aus den Fibroblasten gewonnen wurden. Zudem arbeiten wir auch mit periphären Blutzellen (PBMCs) von Patienten und gesunden Personen.
Unser Fokus liegt auf funktionaler Analyse, welche sich auf Biologie, Biochemie und Physiologie konzentriert. Wir haben einige spezialisierte Methoden etabliert, um mitochondriale Gesundheit zu überwachen und haben kürzlich Methoden in Biochemie, Bildgebung und Durchflusszytometrie für die Untersuchung des endosomalen-lysosomalen Systems entwickelt.

Die Rolle von PINK1 in einem aus iPSCs differenzierten neuronalen Modell von Morbus Parkinson
Anna Schädler, Christine Bus (Alumni)
Wir untersuchen die Rolle der mitochondrialen Kinase PINK1, die eine Rolle in Morbus Parkinson spielt, in humanen, aus iPSCs stammenden dopaminergen Neuronen und nutzen Mechanismen der Geneditierung, um isogene Kontrollen zu generieren. Durch verschiedene etablierte Methoden zur Überprüfung der mitochondrialen Funktion, gelang es uns bestimmte mitochondriale Phänotypen zu identifizieren, die auf eine Rolle für PINK1 in der mitochondrialen Qualitätskontrolle hindeuten. Diese PINK1-defizienten Modelle und ihre isogenen Kontrollen werden wir nun benutzten, um nach genetischen und pharmakologischen Modifikatoren für die Entwicklung neuer neuroprotektiver Strategien zu suchen.
Max Mattheuer
Mutationen der mitochondrialen Kinase PINK1 sind die zweithäufigste Ursache für rezessiv vererbten Morbus Parkinson. Der Mechanismus, welcher zum Absterben von PINK1 mutierten dopaminergen Neurone des Mittelhirns führt, ist bislang ungewiss. Durch die Arbeit an dopaminergen PINK1 KO Neuronen, welche aus iPS Zellen differenziert werden, versuchen wir tiefergehende Einblicke in diese Mechanismen zu erlangen. In PINK1 KO Neuronen konnte keine klassische Mitophagie nach Depolarisierung der mitochondrialen Membran beobachtet werden, während Autophagie und ROS-Produktion unverändert blieben. Das Ziel des Projekts ist es, mögliche Mechanismen der mitochondrialen Qualitätskontrolle / Recycling in PINK1 KO dopaminergen Neuronen in vitro zu finden.
Die Rolle von Miro1 in der mitochondrialen Qualitätskontrolle
Lisa Schwarz
Mitochondrien spielen eine wichtige Rolle in vielen zellulären Vorgängen, wie Energieversorgung, Kalziumgleichgewicht und Zelltod. Im Gegensatz zur Matrix und inneren Membran, welche gut charakterisiert sind, weiß man wenig über die äußere Membran (mitochondrial outer membrame = MOM). Das DFG Graduiertenkolleg (GRK) MOMrane wurde mit dem Ziel gegründet, um die unterschiedlichen Funktionen der äußeren Membran, sowie ihre Struktur, Regulation und Biogenese zu untersuchen. Das GRK besteht aus zehn Doktoranden in verschiedenen Instituten in Tübingen, wovon jeder auf einen anderen Aspekt der Charakterisierung fokussiert ist. Das Projekt wird in Kollaboration mit Partnerlaboren des Weizmann Instituts in Rehovot (Israel) ausgeführt.
Miro1 ist ein Protein der äußeren mitochondrialen Membran und verantwortlich für mitochondrialen Transport. Als Teil des MOMbrane GRK untersuchen wir die Funktion von Miro1 in mitochondrialem Turnover, Metabolismus und Kalziumsignaling. Eine veränderte Funktion der Mitochondrien ist eines der pathologischen Hauptmerkmale von Morbus Parkinson. Wir möchten biologische Rolle von Miro1 untersuchen und zusätzlich seine Relevanz für Morbus Parkinson, indem wir relevante Krankheitsmodelle studieren. Um dieses Verständnis zu erlangen, werden wir krankheits-assoziierte und funktionelle Mutationen in Miro1 in iPSCs mithilfe von Genomeditierung einbringen. Gesunde und editierte iPSCs können dann in andere Zelltypen, wie beispielsweise Neurone, ausdifferenziert werden.
Die Rolle des endosomalen-lysosomalen Systems in atypischem Morbus Parkinson/ Kortikobasalen Syndrom
Katharina Stegen (Alumni)
Die Dysfunktion des endosomalen-lysosomalen Systems ist ein Phänomen das sich durch viele neurodegenerative Erkrankungen, wie Alzheimer, Parkinson und andere seltenere neurodegenerative Erkrankungen wie progressive supranukleäre Blickparese (PSP) und kortikobasales Syndrom/Degeneration (CBS/CBD), zieht. Das endosomale-lysosomale System wird durch Änderungen im pH gebildet und angetrieben, deshalb sind Ionentauscher, die sich in den membranösen Kompartimenten des endosomalen Lumens befinden, kritisch für eine genaue pH-Regulierung. Diese Ionentauscher sind so wichtig, dass große Defekte in ihnen oft von Geburt an eine schwere mentale Retardierung auslösen. Wir hingegen untersuchen Defekte, die die Anfälligkeit eine neurodegenerative Krankheit später im Leben zu entwickeln, erhöhen. Wir untersuchen außerdem die Aspekte des zellulären Transportes, die das endosomale-lysosomale System und Autophagie involvieren, die in Morbus Parkinson betroffen sein könnten.
Mitochondriale Biomarker bei sporadischer Parkinson-Krankheit
Julia Fitzgerald, Gerrit Machetanz, Anne Grünewald (LCSB, Universität Luxemburg)
Sporadische Formen von Morbus Parkinson sind am häufigsten, aber sie sind im Labor schwierig zu modellieren, da keine bestimmte Genmutation bekannt ist, die eingeführt oder korrigiert werden kann, um isogene Kontrollen zu erzeugen. Statistisch gesehen sind große Kohorten erforderlich, um gesunde Individuen mit sporadischen Parkinsonpatienten zu vergleichen. Dies stellt ein Problem dar, da die Kultivierung von Zelllinien, die aus Patientenproben gewonnen werden, parallel für solch große Kohorten spezialisierte Roboter erfordert, für Patienten invasiv ist und sehr teuer ist. Um dies zu überwinden, sammeln wir Blutzellen aus einer großen Kohorte von Parkinsonpatienten einschließlich sporadischer und familiärer Fälle, in denen mitochondriale Proteine betroffen sind. Wir werden nach Änderungen in den Mitochondrien in diesen Zellen suchen, um zu versuchen, Subgruppen von Patienten zu identifizieren, die in Zukunft von Medikamenten profitieren könnten, die auf mitochondriale Dysfunktion abzielen
BEST - Biomarkerevaluation zur Unterstützung der klinischen Translation für Schizophrenien
Richard Wüst, Maria Zarani, Julia Fitzgerald
Psychosen sind eine Gruppe von Erkrankungen von außerordentlicher Heterogenität. Dies hat zur Folge, dass Diagnosen, Prognosen und Behandlungsentscheidungen für einzelne Patienten eine große Herausforderung darstellen. Das Vorhaben optimiert Biomarkersignaturen von Patienten mit hohem Risiko für Schizophrenien und Patienten mit definierten psychotischen Erkrankungen. Dies wird durch Integration von multimodalen Patientendaten und nachfolgender Validierung von Biomarkersignaturen an Patienten-abgeleiteten Neuronen erreicht.
Das durch das BMBF (FKZ: 01EK2101A) geförderte Projekt wird von den 4 Partnern LMU München, ZI Mannheim, NMI Reutlingen und dem Hertie Institut für klinische Hirnfoschung/Universitätsklinik für Psychiatrie Tübingen (HIH/UKPP) getragen. Die Validierung mitochondrialer Parameter erfolgt durch das HIH/UKPP.

+49 (0)7071-
29-87616

+49 (0)7071-
29-87616

07071
29-81971

+49(0)7071-
29-87616
+49 (0)7071-
29-81971

07071
29-87616

+49 (0)7071-
29-87616

2022
Schmidt S, Luecken MD, Trümbach D, Hembach S, Niedermeier KM, Wenck N, Pflügler K, Stautner C, Böttcher A, Lickert H, Ramirez-Suastegui C, Ahmad R, Ziller MJ, Fitzgerald JC, Ruf V, van de Berg WDJ, Jonker AJ, Gasser T, Winner B, Winkler J, Vogt Weisenhorn DM, Giesert F, Theis FJ & Wurst W. Primary cilia and SHH signaling impairments in human and mouse models of Parkinson’s disease. Nat Commun 2022; 13, 4819 PMID: 35974013
Arena G, Sharma K, Agyeah G, Krüger R, Grünewald A, Fitzgerald JC. Neurodegeneration and Neuroinflammation in Parkinson's Disease: a Self-Sustained Loop Curr Neurol Neurosci Rep 2022; Jun 8:1–14 PMID: 35674870
Harmuth T, Weber JJ, Zimmer AJ, Sowa AS, Schmidt J, Fitzgerald JC, Schöls L, Riess O, Hübener-Schmid J. Mitochondrial Dysfunction in Spinocerebellar Ataxia Type 3 Is Linked to VDAC1 Deubiquitination Int. J. Mol. Sci 2022; 23, 5933 PMID: 35682609
Schwarz L and Fitzgerald JC. Steady-State Levels of Miro1 Linked to Phosphorylation at Serine 156 and Mitochondrial Respiration in Dopaminergic Neurons Cells 2022; 11, no. 8: 1269. PMID: 35455950
Kakade P, Ojha H, Raimi OG, Kakade P, Ojha H, Raimi OG, Shaw A, Waddell AD, Ault JR, Burel S, Brockmann K, Kumar A, Ahangar MS, Krysztofinska EM, Macartney T, Bayliss R, Fitzgerald JC, Muqit MMK. Mapping of a N-terminal α-helix domain required for human PINK1 stabilization, Serine228 autophosphorylation and activation in cells. Open Biol. 2022;12(1):210264 PMID: 35042401
Schmidt S, Luecken MD, Trümbach D, Hembach S, Niedermeier KM, Wenck N, Pflügler K, Stautner C, Böttcher A, Lickert H, Ramirez-Suastegui C, Ahmad R, Ziller MJ, Fitzgerald JC, Ruf V, van de Berg WDJ, Jonker AJ, Gasser T, Winner B, Winkler J, Vogt Weisenhorn DM, Giesert F, Theis FJ & Wurst W. Primary cilia and SHH signaling impairments in human and mouse models of Parkinson’s disease. Nat Commun 2022; 13, 4819 PMID: 35974013
Arena G, Sharma K, Agyeah G, Krüger R, Grünewald A, Fitzgerald JC. Neurodegeneration and Neuroinflammation in Parkinson's Disease: a Self-Sustained Loop Curr Neurol Neurosci Rep 2022; Jun 8:1–14 PMID: 35674870
Harmuth T, Weber JJ, Zimmer AJ, Sowa AS, Schmidt J, Fitzgerald JC, Schöls L, Riess O, Hübener-Schmid J. Mitochondrial Dysfunction in Spinocerebellar Ataxia Type 3 Is Linked to VDAC1 Deubiquitination Int. J. Mol. Sci 2022; 23, 5933 PMID: 35682609
Schwarz L and Fitzgerald JC. Steady-State Levels of Miro1 Linked to Phosphorylation at Serine 156 and Mitochondrial Respiration in Dopaminergic Neurons Cells 2022; 11, no. 8: 1269. PMID: 35455950
Kakade P, Ojha H, Raimi OG, Kakade P, Ojha H, Raimi OG, Shaw A, Waddell AD, Ault JR, Burel S, Brockmann K, Kumar A, Ahangar MS, Krysztofinska EM, Macartney T, Bayliss R, Fitzgerald JC, Muqit MMK. Mapping of a N-terminal α-helix domain required for human PINK1 stabilization, Serine228 autophosphorylation and activation in cells. Open Biol. 2022;12(1):210264 PMID: 35042401
2021
Schwarz L, Casadei N and Fitzgerald JC. Generation of R272Q, S156A and K572R RHOT1/Miro1 point mutations in iPSCs from a healthy individual using FACS-assisted CRISPR/Cas9 genome editing. Stem Cell Res. 2021 25(55), 102469. PMID: 34359002
Brown SJ, Boussaad I, Jazaro J, Fitzgerald JC, Antony P, Keatinge M, Blechman, J, Schwamborn J, Krüger R, Placzek M, Bandmann O. PINK1 deficiency impairs adult neurogenesis of dopaminergic neurons. Sci Rep 2021; 11, 6617. PMID: 33758225
Körner A, Bernard A, Fitzgerald JC, Alarcon-Barrerer JC, Kostidis S, Kaussen T, Giera M & Mirakaj V. Sema7A is crucial for resolution of severe inflammation. PNAS 2021; 118, e2017527118. PMID: 33637648
2020
Bus C, Zizmare L, Feldkaemper M, Geisler S, Zarani M, Schaedler A, Klose F, Admard, J, Mageean CJ, Arena J, Fallier-Becker P, Ugun-Klusek A, Maruszczak K, Kapolou K, Schmid B, Rapaport D, Ueffing M, Casadei N, Krüger R, Gasser T, Vogt-Weisenhorn D, Kahle PJ, Trautwein C, Gloeckner CJ and Fitzgerald JC. Human Dopaminergic Neurons Lacking PINK1 Exhibit Disrupted Dopamine Metabolism Related to Vitamin B6 Co-Factors. iScience 2020; 23, 12, 101797. PMID: 33299968
2019
Hertlein V, Flores-Romero H, Das KK, Fischer S, Heunemann M, Calleja-Felipe M, Knafo S, Hipp K, Harter K, Fitzgerald JC, García-Sáez AJ. MERLIN: a novel BRET-based proximity biosensor for studying mitochondria-ER contact sites. Life Sci Alliance 2019; Dec 9;3(1). PMID: 31818884
Grossmann D, Berenguer-Escuder C, Bellet M, Scheibner D, Bohler J, Massart F, Rapaport D, Skupin A, Fouquier d'Hérouël A, Sharma M, Ghelfi J, Raković A, Lichtner P, Antony P, Glaab E, May P, Dimmer K, Fitzgerald JC, Grünewald A, Krüger R. Mutations in RHOT1 Disrupt Endoplasmic Reticulum-Mitochondria Contact Sites Interfering with Calcium Homeostasis and Mitochondrial Dynamics in Parkinson's Disease. Antioxid Redox Signal 2019; Dec 1;31(16):1213-1234. PMID: 31303019
2018
Ugun-Klusek A, Theodosi TS, Fitzgerald JC, Burté F, Ufer C, Boocock D, Yu-Wai-Man P, Bedford L, Billett EE. Monoamine oxidase-A promotes protective autophagy in human SH-SY5Y neuroblastoma cells through Bcl-2 phosphorylation. Redox Biol. 2018; 20,167-181.PMID: 30336354
Jores T, Lawatscheck J, Beke V, Franz-Wachtel M, Yunoki K, Fitzgerald JC, Macek B, Endo T, Kalbacher H, Buchner J, and Rapaport D. Cytosolic Hsp70 and Hsp40 chaperones enable the biogenesis of mitochondrial β-barrel proteins. J. Cell. Biol. 2018; Sep 3;217(9):3091-3108. PMID: 29930205
Fitzgerald JC, Zimprich A, Bobbili DR, May P, Sharma M, Krüger R. Reply: No evidence for rare TRAP1 mutations influencing the risk of idiopathic Parkinson’s disease. Brain 2018; 141 (3):17. PMID: 29373630 * corresponding author
Sofi S, Fitzgerald JC, Jähn D, Dumoulin B, Stehling S, Kuhn H, Ufer C. Functional characterization of naturally occurring genetic variations of the human Guanine-rich RNA sequence binding factor 1 (GRSF1). Biochim. Biophys. Acta. 2018; 1862(4):866-876. PMID: 29366917
Marrone L, Bus C, Schöndorf D, Fitzgerald JC, Kübler M, et al. Generation of iPSCs carrying a common LRRK2 risk allele for in vitro modeling of idiopathic Parkinson's disease. PLOS One 2018; 13(3). PMID: 29513666
Rotermund C, Machetanz G, Fitzgerald JC. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Frontiers in Endocrinology 2018; 19;9:400. PMID:Studenten
PhD
Lisa Schwarz, University of Tübingen (2018-), Katharina Stegen, University of Tübingen (2013-2018), Christine Bus, University of Tübingen (with Thomas Gasser, 2013-2018).
MSc Thesis
Lara-Sophie Rieder, University of Tübingen (2020), Orchid Ammar, University of Tübingen (2020), Kim Krieg, University of Constance (2018-2019), Max Mattheuer, MCBI, University of Tübingen (2018), Anna Schaedler, GTC, Tübingen (2018), Konstantina Kopolou, GTC, Tübingen (2017), Dilara Halim, GTC, Tübingen (2017), Benedikt Hoelbling, GTC, Tübingen (2017), Marco Siekmann, GTC, Tübingen (2017), Lisa Schwarz, GTC, Tübingen (2017).
BSc
Kevin Schindler, University of Tübingen (2015), Rahel Lewin, University of Tübingen (2014)

Hertie-Zentrum für Neurologie
Hertie-Institut für klinische Hirnforschung
Abteilung Neurologie mit Schwerpunkt neurodegenerative Erkrankungen
Otfried Mueller Strasse 27
72076 Tuebingen
Tel.: +49 (0)7071 29 81971
Fax: +49 (0)7071 29-4255




