

Neurogenetische Erkrankungen wie cerebelläre Ataxien, hereditäre spastische Spinalparalysen und Leukodystrophien sind seltene Erkrankungen, für die vielfach die genetische Ursache bis heute ungeklärt ist. Unsere Gruppe bemüht sich krankheitsverursachende Mutationen aufzudecken. Damit erhalten die Patienten nicht nur eine gesicherte Diagnose, sondern erhalten wir auch die Möglichkeit, die Erkrankungen von ihrer Ursache her zu erforschen und über die Aufdeckung der Krankheitsmechanismen nach neuen therapeutischen Ansätzen zu suchen. Für unsere Forschung verwenden wir induzierte pluripotente Stammzellen, die wir in Neurone differenzieren können, so dass wir ein Zellmodel der Erkrankung erhalten, das genetisch identisch ist mit den Zellen, die bei unseren Patienten erkranken.
Im Jahr 2010 wurde von uns in Tübingen das erste Zentrum für Seltene Erkrankungen (ZSE) in Deutschland gegründet mit Unterstützung durch Eva Luise Köhler, der Frau des früheren Bundespräsidenten. Das ZSE hat sich der translationalen Forschung verschrieben. In den Spezialambulanzen werden viele Patienten mit erblichen Bewegungsstörungen betreut und umfangreich standardisiert untersucht. So können wir repräsentative Kohorten dieser seltenen Erkrankungen aufbauen, Daten zur natürlichen Progression erarbeiten, Biomaterial für die Entwicklung von Biomarkern sammeln und damit Therapiestudien optimal vorbereiten.
In der Neurologischen Klinik bieten wir Spezialambulanzen für die Diagnostik und Therapie seltener erblicher Bewegungsstörungen an:
- Zentrum für seltene neurologische Erkrankungen (Chorea Huntington, Mitochondriopathien, etc.)
Unsere klinische Forschung zielt darauf ab, neue Therapien für erbliche Bewegungsstörungen zu entwickeln:
- Der erste Schritt auf diesem Weg ist die Schaffung repräsentativer Patientenkohorten für diese seltenen Erkrankungen. Mit Unterstützung der HSP-Selbsthilfeorganisationen etablieren wir ein Internet-basiertes Register für Hereditäre Spastische Spinalparalysen mit einer standardisierten Erhebung klinischer und genetischer Daten. Die Ergebnisse der Querschnittsuntersuchung der HSP-Kohorte konnten jüngst publiziert werden (Schüle et al., 2016). Die Längsschnittuntersuchung wird prospektive Daten zur Krankheitsprogression liefern. Solche Daten sind eine elementare Voraussetzung für die Planung von Therapiestudien, die darauf abzielen, den natürlichen Erkrankungsverlauf zu verlangsamen oder gar zu stoppen. Ähnliche Studien führen wir auch für die Friedreich Ataxie und andere rezessiv vererbte Ataxien, spinocerebelläre Ataxien, die Multisystematrophie und adulte Verlaufsformen der Leukodystrophien durch.
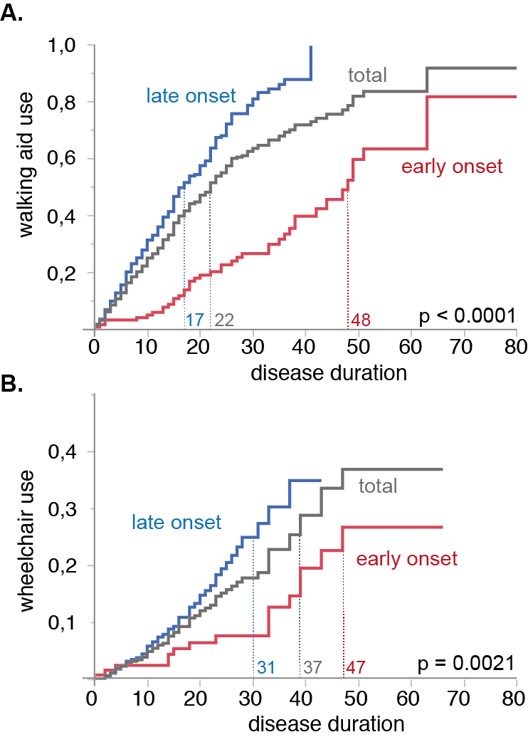
- Da die klinische Progression bei den meisten Erkrankungen relativ langsam ist und von vielen externen Faktoren abhängt, suchen wir nach Biomarkern, die die Krankheitsaktivität widerspiegeln. Für die hereditäre diffuse Leukencephalopathie mit axonalen Sphäroiden (HDLS) erstellen wir z.B. Cytokinprofile im Blut und Liquor auf der Suche nach Markern, die in der Pathogenese der HDLS involviert sind. Analog entwickeln wir auch Biomarker, die die Progression bei Ataxien und HSP anzeigen können.
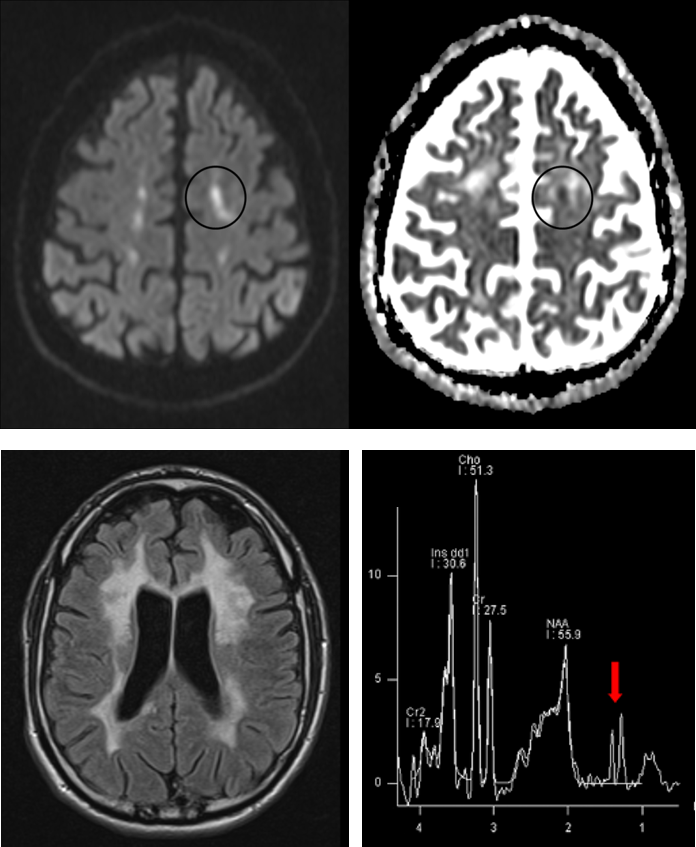
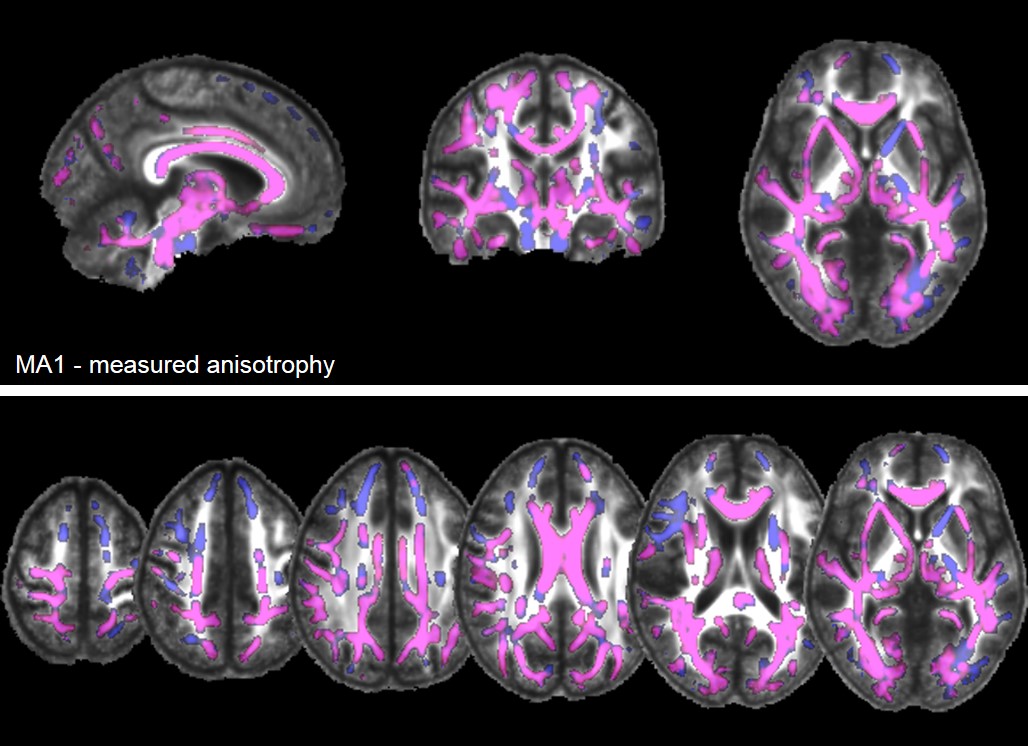
- 3. Idealerweise würden Therapien einsetzen, bevor Symptome der erblichen Erkrankungen auftreten, und könnten so den Erkrankungsbeginn verzögern oder gar verhindern. Bei autosomal dominant vererbten Erkrankungen wie der HDLS, der SPG4 oder den spinocerebellären Ataxien (SCA) haben erstgradige Verwandte von Patienten ein 50% Risiko, die Mutation geerbt zu haben. Durch sehr sorgfältige Untersuchungen solcher Risikopersonen versuchen wir Marker zu finden, die Erkrankungsaktivität in der präsymptomatischen Phase der Erkrankung anzeigen, also bevor die Patienten Beschwerden bekommen. Studien zur Etablierung präsymptomatischer Biomarker führen wir bei gesunden Mutationsträgern der HDLS, der SPG4 (preSPG4 study) und der spinocerebellären Ataxien durch (RISCA study; Jacobi et al., 2015) unter Einsatz von computerisierter Bewegungsanalyse, neuen Kernspin- / MRT-Bildgebungstechniken und Bestimmungen der mutierten Krankheitsproteine (Lindig et al., 2015).
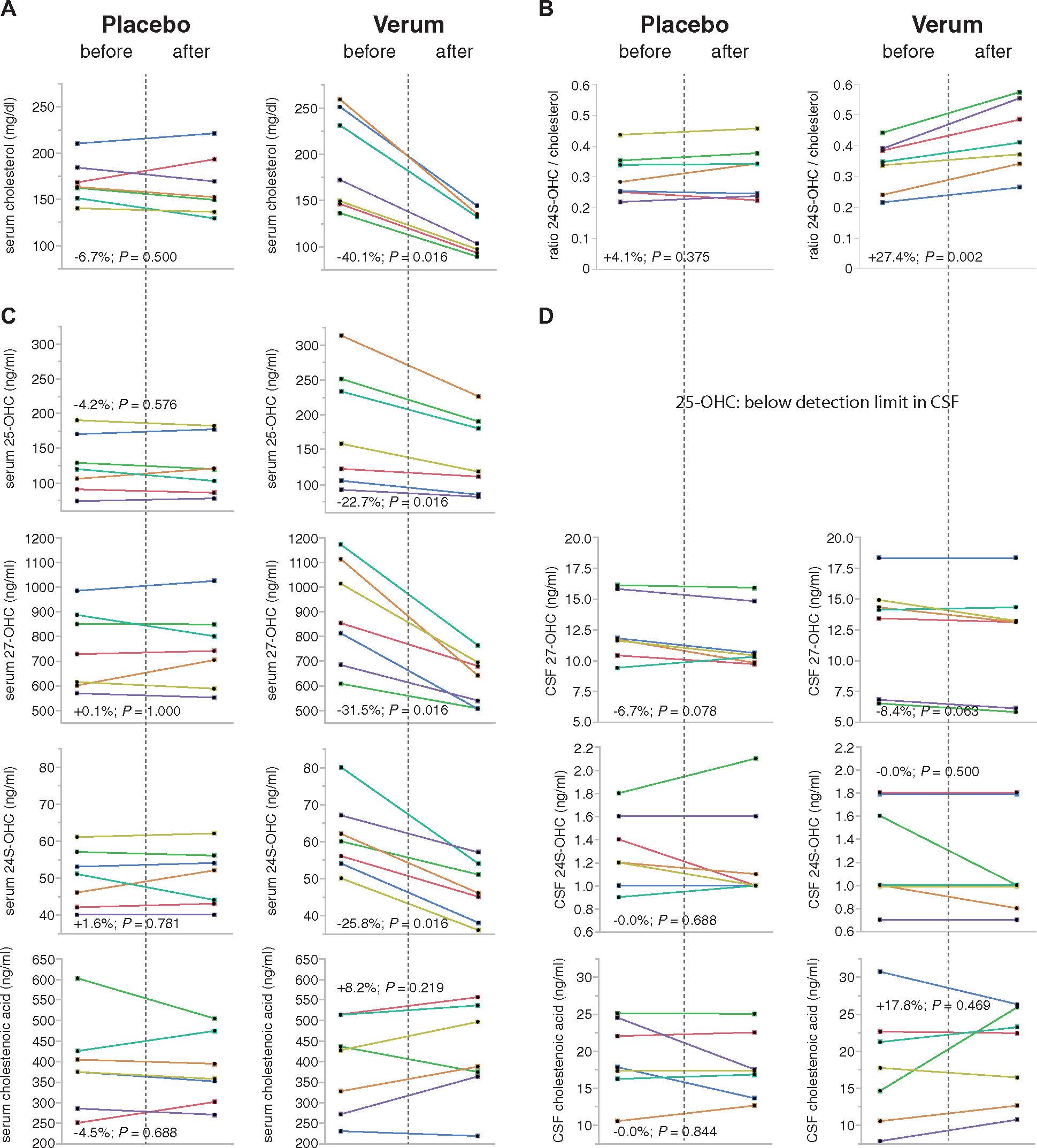
- Das eigentliche Ziel dieser Forschungsanstrengungen ist die Ermöglichung von Therapiestudien mit neuen Substanzen, die in den Krankheitsprozess eingreifen. Auf die Entwicklung solcher Therapieansätze zielt auch die experimentelle Forschung in unserem Labor ab. Ein Beispiel für diesen translationalen Forschungsansatz ist die erste randomisierte, Placebo kontrollierte Studie bei der hereditären spastischen Spinalparalyse Typ 5 (SPG5) (Schöls et al., 2017).
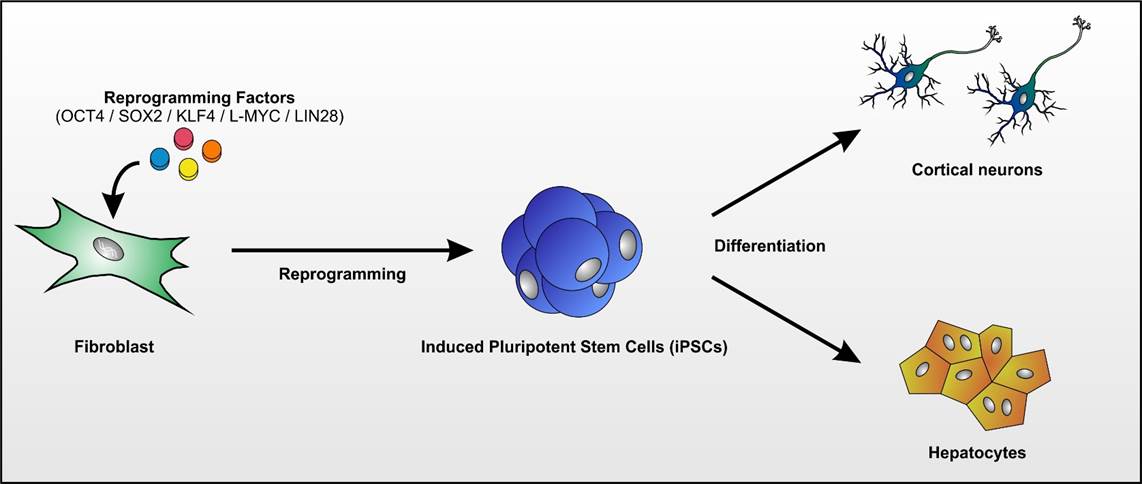
Das Labor der AG Schöls profitiert von dem unmittelbaren Zugang zu Biomaterialien von Patienten-mit seltenen neurogenetischen Erkrankungen und ist auf die Aufdeckung neuer Krankheitsgene und die Untersuchung der Erkrankungsmechanismen in induzierten pluripotenten Stammzellen (iPSC) fokussiert.
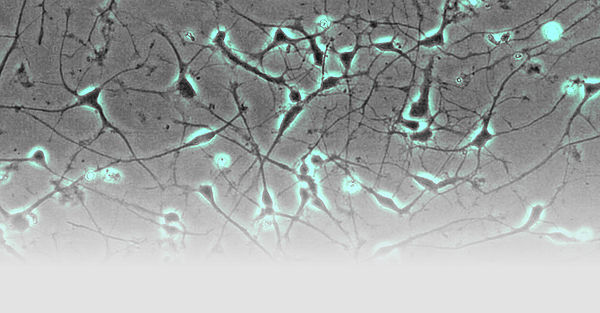
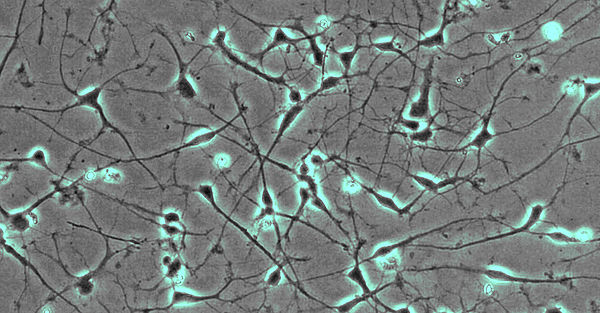
- Fibroblasten gewonnen aus Hautbiopsien werden mittels Nukleofektion episomaler Plasmide mit den klassischen Reprogrammierungsfaktoren in iPSCs umgewandelt (Abb. 1; Hauser et al., 2016). Diese Stammzellen können dann wiederum in Neurone oder andere für die Pathogenese relevante Gewebe (wie z.B. Hepatozyten) re-differenziert werden. Hierdurch wird es möglich, Neurone in der Kulturschale zu untersuchen, die mit unseren Patienten genetisch identisch sind.
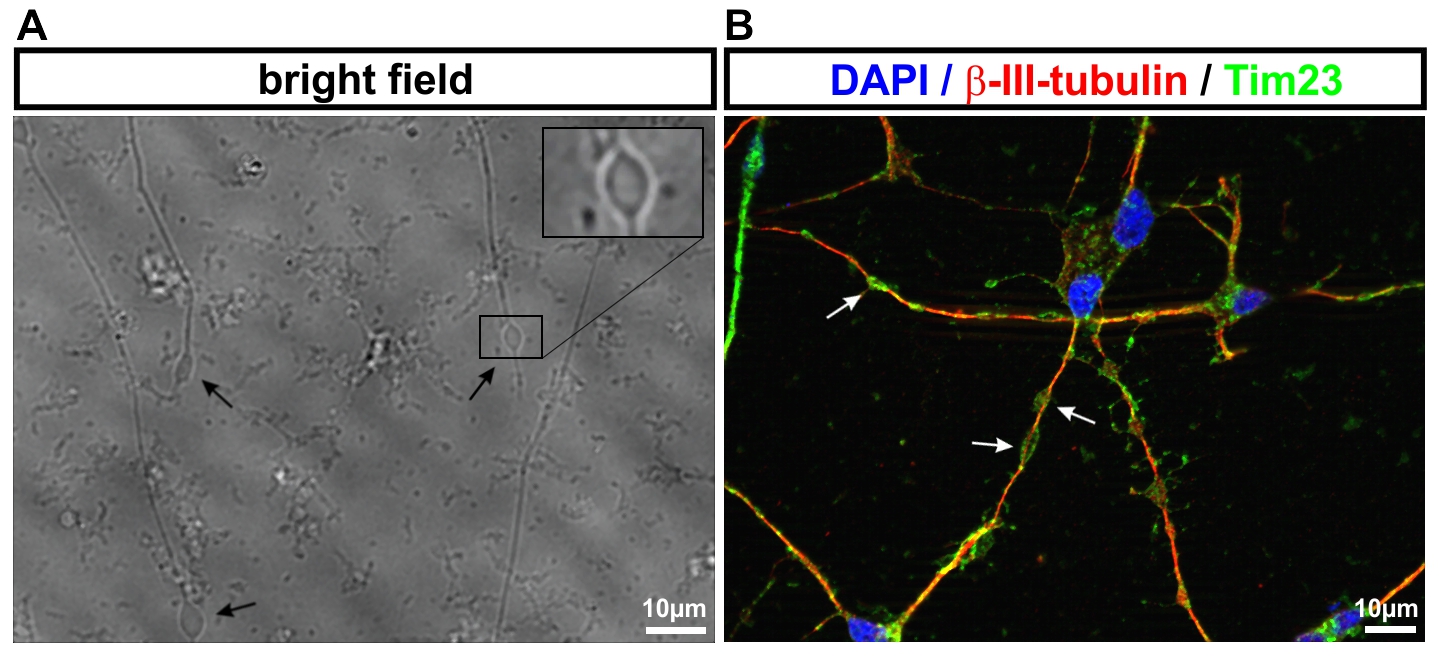
- Neurone aus iPS-Zellen ermöglichen es uns, die jeweiligen Erkrankungsprozesse nachzubilden. So werden von der hereditären spastischen Spinalparalyse (HSP) schwerpunktmäßig die langen Axone des corticospinalen Trakts betroffen. Mit Hilfe des live cell imaging und des high content imaging konnten wir in der Kulturschale zeigen, dass das Axonwachstum bei Neuronen von SPG4-Patienten (der häufigsten Form der HSP) eingeschränkt ist. Darüber hinaus entwickeln SPG4-Neurone vermehrt Axonschwellungen, in denen Zellfragmente abgelagert sind, die den axonalen Transport und damit die Funktion der Nervenzelle stören.
- Die SPG5 ist ein Subtyp der HSP, der durch Mutationen im Cytochrom CYP7B1 verursacht wird. Bei SPG5-Patienten führt der Mangel an CYP7B1 in der Leber zu metabolischen Veränderungen und einem exzessiven Anstieg von Oxysterolen im Blut aber auch im Liquor. Werden Neurone aus iPS-Zellen mit Oxysterolen in Konzentrationen kultiviert, wie wir sie bei SPG5-Patienten finden, hemmt dies das Axonwachstum und auch das Überleben der Nervenzellen. Oxysterole sind daher gute Biomarker für die SPG5. Aktuell untersuchen wir die Oxysterolprofile in Hepatozyten, die wir aus iPS-Zellen von SPG5 Patienten differenziert haben, mit Hilfe der Massenspektrometrie in Kooperation mit Prof. Ingemar Björkhem (Karolinska Institut, Stockholm) und Prof. William Griffiths (Swansea, UK).
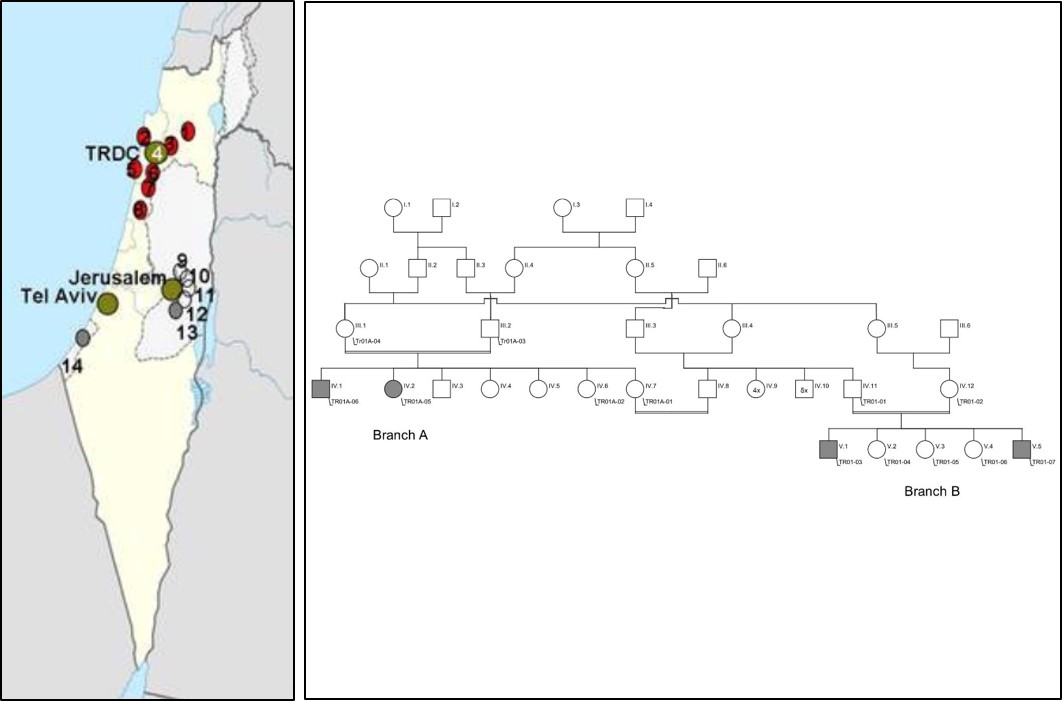
- Mit Hilfe moderner Sequenziertechniken suchen wir nach den genetischen Ursachen bei Patienten mit ultra-seltenen Erkrankungen. Die DFG fördert ein von uns geleitetes trilaterales Projekt, in dem israelische, palästinensische und deutsche Wissenschaftler nach neuen Genen in Familien mit bislang unklaren Krankheiten suchen. Genetische Erkrankungen sind in Israel und Palästina aufgrund der weit verbreiteten Konsanguinität häufig. Mittels Exomsequenzierung konnten wir etliche neue Krankheitgene identifizieren wie AP4B1, WWOX and DNAJC3 (Bauer et al. 2012; Mallaret et al. 2014; Synofzik et al. 2014). Whole genome Analysen führen wir zur Aufdeckung neuer Mutationstypen wie intronischer Mutationen in europäischen Verbundprojekten wie NEUROMICS in enger Kooperation mit Prof. Olaf Rieß (Medizinische Genetik, Tübingen), Prof. Michel Koenig (Montpellier), Dr. Holger Prokisch (Helmholtz Zentrum, München) and Prof. Stephan Züchner (Hussman Institute, Miami) durch.


+49 (0)7071-
29-85653

+49(0)7071-
29-85170

+49 (0)7071-
29-87654

+49(0)7071-
29-85247

+49(0)7071-
9254-402

+49(0)7071-
9254-402

+49(0)7071-
29-85247

+49 (0)7071-
29-81969

+49(0)7071-
9254-054

+49(0)7071-
29-87609

+49(0)7071-
29-85374

+49(0)7071-
29-85247

+49(0)7071-
29-87609

+49(0)7071-
29-87609

+49 (0)7071-
29-85247

+49(0)7071-
29-82057

+49 (0)7071-
29-85374

+49(0)7071-
29-82057

+49 (0)7071-
29-85247

+49(0)7071-
29-85247

+49(0)7071-
9254-054

+49 (0)7071-
29-85247

+49(0)7071-
29-85247
Gesamtübersicht der Publikationen
Ausgewählte Publikationen
Hengel H, Hannan SB, Dyack S, …, Schuster S, Hauser S, Admard J, Casadei N, Velic A, Macek B, Ossowski S, Houlden H, Maroofian R, Schöls L. Bi-allelic loss-of-function variants in BCAS3 cause a syndromic neurodevelopmental disorder. Am J Hum Genet. 2021;108:1069-1082
Collier JJ, Guissart C, Oláhová M, …, Schöls L, … Taylor RW. Developmental consequences of defective ATG7-mediated autophagy in humans. N Engl J Med. 2021; 384:2406-2417
Hengel H, Bosso-Lefèvre C, Grady G, … , Schöls L#, Reversade B#. Loss-of-function mutation in UDP-Glucose-6-Dehydrogenase cause recessive developmental epileptic encephalopathy. Nat Comm. 2020;11:595
Hauser S, Schuster S, Heuten E, Hoeflinger P, Admard J, Schelling Y, Velic A, Macek B, Ossowski S, Schöls L. Comparative Transcriptional Profiling of Motor Neuron Disorder-Associated Genes in Various Human Cell Culture Models. Frontiers Cell Dev Biol. 2020; ;8:544043
Synofzik M, Puccio H, Mochel F, Schöls L. Autosomal recessive cerebellar ataxias: paving the way towards targeted molecular therapies. Neuron. 2019;101:560-83
Hauser S, Poenisch M, Schelling Y, Hoflinger P, Schuster S, Teegler A, Betten R, Gustafsson JA, Hubener-Schmid J, Schlake T, Chevessier-Tunnesen F, Horscroft N, Bjorkhem I, Schöls L. mRNA as a Novel Treatment Strategy for Hereditary Spastic Paraplegia Type 5. Mol Ther Methods Clin Dev. 2019;15:359-370
Diallo A, Jacobi H, Cook A, Labrum R, Durr A, Brice A, Charles P, Marelli C, Mariotti C, Nanetti L, Panzeri M, Rakowicz M, Sobanska A, Sulek A, Schmitz-Hubsch T, Schöls L, Hengel H, Melegh B, Filla A, Antenora A, Infante J, Berciano J, van de Warrenburg BP, Timmann D, Boesch S, Pandolfo M, Schulz JB, Bauer P, Giunti P, Kang JS, Klockgether T, du Montcel ST. Survival in patients with spinocerebellar ataxia types 1, 2, 3, and 6 (EUROSCA): a longitudinal cohort study. Lancet Neurol. 2018;17:327-34
Schöls L, Rattay TW, Martus P, Meisner C, Baets J, Fischer I, Jagle C, Fraidakis MJ, Martinuzzi A, Saute JA, Scarlato M, Antenora A, Stendel C, Hoflinger P, Lourenco CM, Abreu L, Smets K, Paucar M, Deconinck T, Bis DM, Wiethoff S, Bauer P, Arnoldi A, Marques W, Jardim LB, Hauser S, Criscuolo C, Filla A, Zuchner S, Bassi MT, Klopstock T, De Jonghe P, Bjorkhem I, Schule R. Hereditary spastic paraplegia type 5: natural history, biomarkers and a randomized controlled trial. Brain. 2017;140:3112-7
Schüle R, Wiethoff S, Martus P, Karle KN, Otto S, Klebe S, Klimpe S, Gallenmüller C, Kurzwelly D, Henkel D, Rimmele F, Stolze H, Kohl Z, Kassubek J, Klockgether T, Vielhaber S, Kamm C, Klopstock T, Bauer P, Züchner S, Liepelt-Scarfone I, Schöls L. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann Neurol. 2016;79:646-58
Schöls L, Reimold M, Seidel K, Globas C, Brockmann K, Hauser TK, Auburger G, Bürk K, den Dunnen W, Reischl G, Korf HW, Brunt ER Rüb U. No parkinsonism in SCA2 and SCA3 despite severe neurodegeneration of the dopaminergic substantia nigra. Brain. 2015;138:3316-26

Hertie-Zentrum für Neurologie
Hertie-Institut für klinische Hirnforschung
Abteilung Neurologie mit Schwerpunkt neurodegenerative Erkrankungen
Hoppe-Seyler-Straße 3
72076 Tübingen
Tel.: +49 (0)7071 29-82057
Fax: +49 (0)7071 29-4254




