

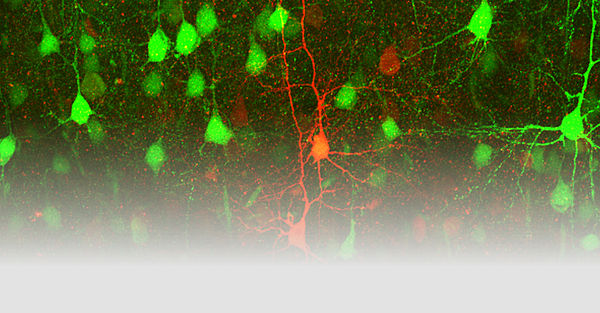
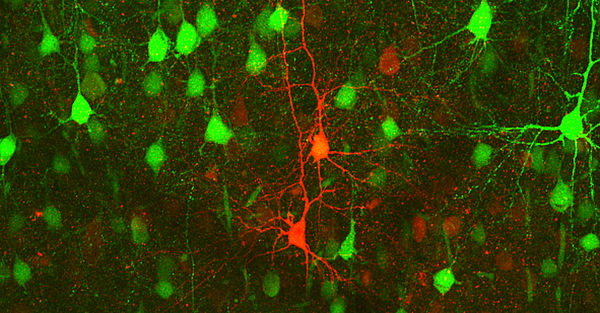
Unsere Arbeitsgruppe untersucht mit Hilfe transgener Mausmodelle die molekularen Mechanismen krankheitsassoziierter Mutationen in Genen, welche neuronale Ionenkanäle kodieren. Zu diesen sogenannten Kanalopathien zählen auch Epilepsien und Migräne, zwei der häufigsten neurologischen Erkrankungen.
Epileptische Anfälle resultieren aus einer spontanen, synchronen elektrischen Aktivität von Neuronengruppen im Gehirn, die sich in einem breiten Spektrum klinischer Symptome äußern, wie etwa einem Kribbeln in einer Extremität bis hin zu Bewusstseinsstörungen, komplexen Handlungen oder generalisierten Anfällen. Migräneattacken beginnen oft mit einer Aura, die sich klassischerweise in Sehbeschwerden äußert. Diese Auren sind auf eine kortikale Streudepolarisation (CSD) zurückzuführen, eine Welle neuronaler Depolarisation, die langsam über die Großhirnrinde läuft. Beide Krankheiten sind also mit einer pathologischen elektrischen Aktivität verbunden, Epilepsie mit einer schnellen, die Migräne mit einer langsamen Ausbreitung der abnormen Aktivität. Interessanterweise können Defekte im selben Gen Epilepsie und Migräne verursachen, beide Erkrankungen können außerdem im selben Patienten auftreten und sich gegenseitig auslösen.
In unseren Projekten wollen wir den Zusammenhang von genetischen Mutationen und seltenen Ionenkanalerkrankungen auf molekularer, zellulärer und Netzwerkebene und den zu Grunde liegenden Mechanismus der Übererregbarkeit dieser Erkrankungen (epileptische Anfälle vs. CSD) verstehen, um neue Therapien entwickeln zu können. Hierfür nutzen wir verschiedene elektrophysiologische und optische Methoden, welche durch Transkriptom-Untersuchungen ergänzt werden. Die seltenen Ionenkanalerkrankungen bieten dabei auch ein Modell für die Pathophysiologie und Therapie von sehr viel häufiger auftretenden, sporadischen Erkrankungen mit ähnlicher Symptomatik.
Epileptogenese von genetischen Epilepsien
Ziel der Forschungsgruppe FOR-2715 „Epileptogenese von genetischen Epilepsien“ ist es zu klären, ob und wie genetische Varianten eine Kaskade epileptogener Ereignisse auslösen und wie diese mit der Gehirnentwicklung interagieren. In unserem Projekt wollen wir die epileptogenen Mechanismen KCNA2-assoziierter Enzephalopathien verstehen. Dafür nutzen wir in utero Elektroporation von KCNA2 Varianten sowie neu entwickelte Knock-in Mausmodelle mit gain- oder loss-of-function Varianten. Mit Hilfe dieser Mausmodelle wollen wir das vulnerable Zeitfenster von Kcna2-Varianten und die Netzwerkdysfunktion des sich entwickelnden neuronalen Netzwerks untersuchen. Zusätzlich beschäftigen wir uns mit der Behandlung von Patienten mit KCNA2-gain-of-function Varianten mit 4-Aminopyridin und entwickeln derzeit Kcna2-Antisense-Oligonukleotide als neue Therapiemöglichkeit.
Treat-ION
Im Forschungsverbund Treat-ION konzentrieren wir uns auf die pathophysiologischen Mechanismen der hemiplegischen Migräne (HM), einer schweren monogenen Unterform der Migräne mit einem gewissen Grad an halbseitiger motorischer Schwäche während der Aura. Dabei wollen wir die pathophysiologischen Mechanismen epileptischer Anfälle und kortikaler Streudepolarisation untersuchen, welche durch Varianten im SLC1A3 Gen verursacht werden, die den Glutamat-Transporter und Anionenkanal EAAT1 kodieren. Zusätzlich wollen wir neue und individualisierte Therapien für Patienten mit dieser seltenen Erkrankung entwickeln.
Gruppo Famiglie Dravet Associazione ONLUS
In einem weiteren Projekt, das von der Gruppo Famiglie Dravet Associazione ONLUS in Partnerschaft mit anderen europäischen Dravet-Stiftungen finanziert wird, interessieren wir uns für einen neuen Mechanismus in der Pathophysiologie des Dravet-Syndroms (DS), nämlich die Interaktionen zwischen Neuronen und Oligodendrozyten. Diese Erkrankung wird meist durch genetische Varianten im SCN1A Gen verursacht, welches den NaV1.1-Kanal kodiert, den wichtigsten Na+-Kanal in hemmenden Neuronen. Hier untersuchen wir die Auswirkungen von SCN1A-Varianten in Neuronen, Oligodendrozyten-Vorläuferzellen (OPCs) und Oligodendrozyten, da diese Zellen alle den NaV1.1-Kanal exprimieren, einen gemeinsamen embryonalen Ursprung haben und während der Entwicklung stark interagieren. Zusätzlich wollen wir die morphologischen Veränderungen des Axoninitialsegments und der Axone identifizieren und die Myelinisierung in DS-Mausmodelle untersuchen
European Joint Program on Rare Disease
Im Rahmen des European Joint Program on Rare Disease (EJP RD) konzentrieren wir uns als Teil des national geförderten (von der DFG für Deutschland) Konsortiums SCN1A-UP! mit Partnern in Frankreich, Italien, Belgien, den Niederlanden und Deutschland auf gezielte Behandlungen des Dravet-Syndroms unter Verwendung von Knock-in-Tiermodellen sowie von menschlichen Krankheitsmodellen.

+49 (0) 7071
29-81984

+49 (0) 7071
2981914

+49 (0) 7071
29-80440

+49 (0) 7071
29- 81968
+49 (0)7071
29-81921
Die vollständige Publikationsliste finden Sie auf ORCID
Hedrich UBS*, Lauxmann S*, Wolff M, …, Schwarz N, Fudali M, Lerche H. Effective precision therapy in KCNA2-related developmental and epileptic encephalopathy with 4-aminopyridine, Sci Trans Med 2021 Sep;13(609):eaaz4957, *equal contribution.
Auffenberg E*, Hedrich UBS*, Barbieri R*, Miely D*, Groschup B, Wuttke TV, Vogel N, Lührs P, Zanardi I, Bertelli S, Spielmann N, Gailus-Durner V, Fuchs H, Hrabe de Angelis M, Pusch M, Dichgans M, Lerche H, Gavazzo P§, Plesnila N§, Freilinger T§. Hyperexcitable interneurons trigger cortical spreading depression in an Scn1a migraine-model, JCI 2021 Nov 1;131(21):e142202. *equal contribution.
Lauxmann S, Verbeek NE, Liu Y, Zaichuk M, Müller S, Lemke JR, van Kempen MJA, Lerche H, Hedrich UBS. Relationship of electrophysiological dysfunction and clinical severity in SCN2A-related epilepsies. Hum Mutat 2018;39:1942-1956.
Masnada S*, Hedrich UBS*, Gardella E, Schubert J, Kaiwar C, Klee EW, Lanpher BC, Gavrilova RH, Synofzik M, Bast T, …, Lerche H, Rubboli G. Clinical spectrum and genotype–phenotype associations of KCNA2-related encephalopathies. Brain 2017;140(9):2337-54, *equal contribution.
Wolff M*, Johannesen KM*, Hedrich UB*, Masnada S, Rubboli G, Gardella E, Lesca G, Ville D, Milh M, Villard L, Afenjar A, Chantot-Bastaraud S, Mignot C, Lardennois C, Nava C, Schwarz N, …, Kluger G, Lerche H & Moller RS. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 2017; * equal contribution.
Schwarz N, Hahn A, Bast T, Muller S, Loffler H, Maljevic S, Gaily E, Prehl I, Biskup S, Joensuu T, Lehesjoki AE, Neubauer BA, Lerche H & Hedrich UB. Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J Neurol 2016;263:334-43.
Syrbe S*, Hedrich UB*, Riesch E*, Djemie T*, Muller S, Moller RS, Maher B, Hernandez-Hernandez L, Synofzik M, Caglayan HS, Arslan M, Serratosa JM, Nothnagel M, May P, Krause R, Loffler H, Detert K, Dorn T, Vogt H, Kramer G, Schols L, Mullis PE, Linnankivi T, Lehesjoki AE, Sterbova K, Craiu DC, Hoffman-Zacharska D, Korff CM, Weber YG, Steinlin M, Gallati S, Bertsche A, Bernhard MK, Merkenschlager A, Kiess W, Euro ER, Gonzalez M, Zuchner S, Palotie A, Suls A, De Jonghe P, Helbig I, Biskup S, Wolff M, Maljevic S, Schule R, Sisodiya SM, Weckhuysen S, Lerche H, Lemke JR. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat Genet 2015;47:393-9 * equal contribution.
Hedrich UBS, Liautard C, Kirschenbaum D, Pofahl M, Lavigne J, Liu Y, Theiss S, Slotta J, Escayg A, Dihne´M, Beck H, Mantegazza M, and Lerche H. Impaired Action Potential Initiation in GABAergic Interneurons Causes Hyperexcitable Networks in an Epileptic Mouse Model Carrying a Human NaV1.1

Hertie-Zentrum für Neurologie
Hertie-Institut für klinische Hirnforschung
Abteilung Neurologie mit Schwerpunkt Epileptologie
Otfried-Müller-Str. 27
72076 Tübingen
Tel.: +49 (0)7071 29 81984
Fax: +49 (0)7071 29-4698




